Algorithms Overview
barcode/UMI extraction
SeekSoulTools is able to extract the barcode and UMI sequences based on different Read1 structures. The pipeline processes the barcodes and filters Read1 and its corresponding Read2, and an updated fastq file is generated at the end of step 1.
Structure design and description
Barcode and UMI descriptions: Describing the basic structure of Read1 using letters and numbers, where the letters represent the meaning of the nucleotides and the numbers represent the length of the nucleotides.
B: barcode bases
L: linker bases
U: UMI bases
X: other arbitrary bases used as placeholders
The Read1 structure of the SeekOne™ DD Single Cell Immune Profiling Kit is B17U12:

Workflow
Barcode and linker correction
After determining the starting position of the barcode, the corresponding sequence is extracted based on the structural design. When the extracted barcode sequence is found in the whitelist, it is considered a valid barcode and the read count with valid barcodes is recorded. When the barcode is not found in the whitelist, it is considered an invalid barcode
During sequencing process, there is a certain probability that sequencing error occurs. With the presense of a whitelist, SeekSoulTools are able to do barcode correction. When correction is enabled, if sequences that are one base differ (one humming distance apart) from the invalid barcode appear in the whitelist, we will consider the following circumstances:
If there is one and only sequence exists in the whitelist, we will correct the invalid barcode to the barcode in the whitelist.
If multiple sequences exist in the whitelist, we will correct the invalid barcode to the sequence with the highest read support.
Adaptors and polyA sequence trimming
In transcriptome, polyA tail and Adapter sequences introduced during library preparation, may appear at the end of Read2.We remove these contaminating sequences and make sure the trimmed Read2 length is greater than the minimum length for accurate alignment afterward. If the trimmed Read2 length is less than the minimum length, we consider the read to be invalid.
After the processing procedure described above, the metrics is shown below:
total: Total reads
valid: Number of reads without correction and number of successfully corrected reads
B_corrected: Number of successfully corrected reads
B_no_correction: Number of reads with incorrect barcodes
trimmed: Number of reads that have been trimmed
too_short: Number of reads that are shorter than 60bps after trimming
Number of reads in the updated FASTQ file: total_output = valid - too_short
Sequence Assembly
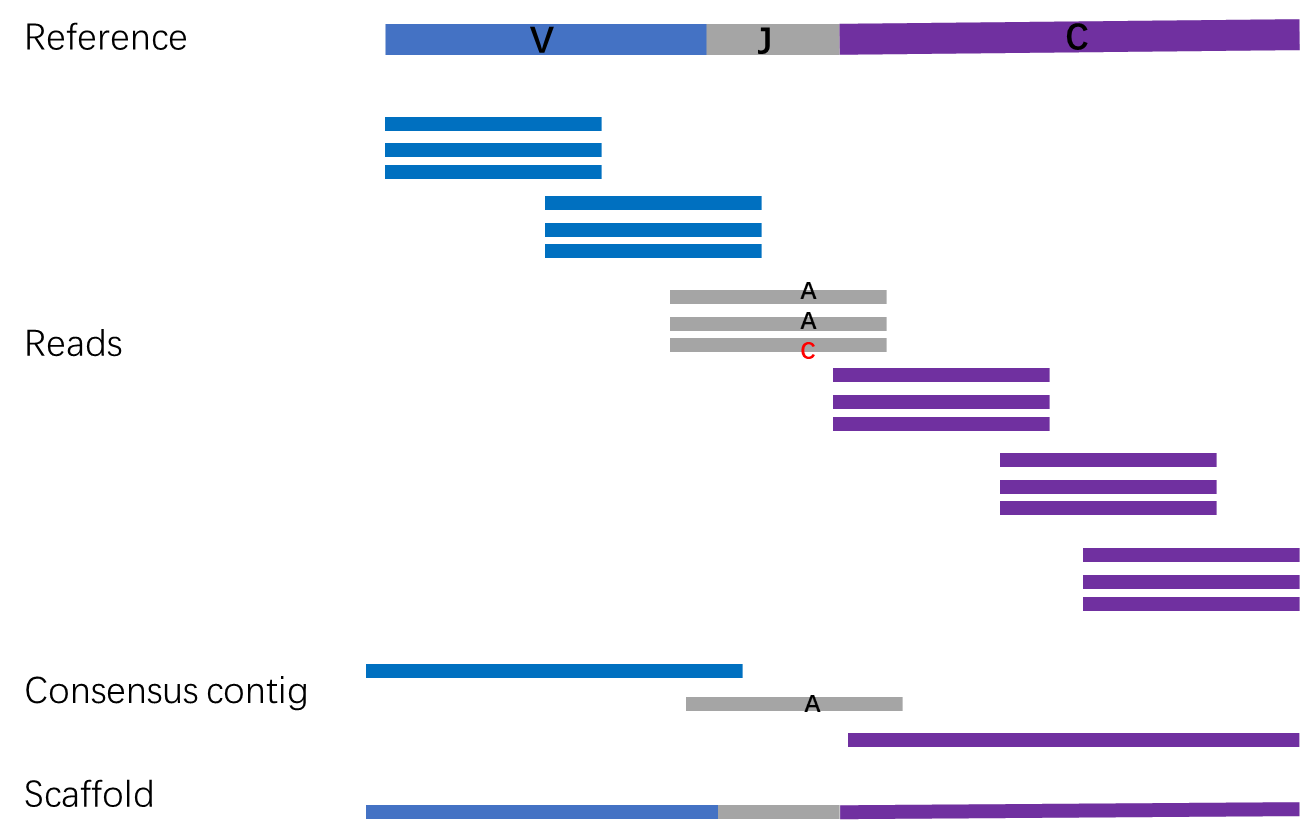
VDJ sequence assembly refers to the process of assembling short sequencing reads into longer VDJ sequences. It primarily involves the following steps (as illustrated above):
Aligning reads to a reference sequence to obtain their coordinates relative to the reference sequence.
First, the reads and VDJ germline reference sequences are separately divided into 11-base-pair kmers. Subsequently, the number of shared kmers between the read and reference sequence is computed as the length of the alignment between the read and the reference sequence. Then, if there is a 22-base-pair region with a perfect match between the read and the reference sequence, the read is considered aligned to the reference sequence.For the read that aligns to the germline reference sequence obtained as described above, we determine its corresponding region (V-REGION, J-REGION, or C-REGION) based on the alignment length.A single read can align to multiple genes within the V-REGION (J-REGION or C-REGION). We align all reads with the same barcode and UMI to the gene within the V-REGION (J-REGION or C-REGION) that has the highest alignment count. This gene is selected as the final reference sequence.Finally, using the reference sequence, each base on the read is annotated with its coordinate relative to the reference sequence.
Correct the bases on the reads that share the same reference sequence coordinates.
Calculate the consistency of bases on the reads at each position corresponding to the reference sequence. If the coverage depth of the reads is greater than or equal to 2 and the base consistency is greater than or equal to 51%, select the base with the highest occurrence. Otherwise, mark it as N.
Assemble the corrected reads into a long consensus contig.
Using the coordinates relative to the V-REGION, J-REGION, or C-REGION, connect the reads together and assemble them into a long consensus contig.There are two methods for assembling the consensus contig (the default option is to use the second method):The first method involves assembling reads with the same UMI. Each UMI results in a contig for the V-REGION, J-REGION, or C-REGION.The second method involves merging all reads from the same V-REGION (J-REGION or C-REGION) gene and obtaining a separate contig for the V-REGION, J-REGION, or C-REGION.
Assemble the consensus contigs of the V-REGION, J-REGION, and C-REGION.
Connect the consensus contigs of the V-REGION, J-REGION, and C-REGION using overlaps to form a complete sequence. Connect contigs that have overlaps of 18 bp or more with each other.
Sequence filtering
Filtering based on assembly metrics:
For contigs that contain both light chain and heavy chain in the barcode, contigs that meet either of the following conditions will be discarded: For BCR, if the contig's UMI is less than 3; for TCR, no such filtering will be applied.
In theory, within a sample, the number of light chains matched to the same heavy chain is limited. However, if contamination occurs, it can result in one heavy chain corresponding to multiple light chains. For contigs with a UMI less than or equal to 10, if they meet either of the following conditions, they will be filtered out: Condition 1 - In all barcodes, if the number of light chain types paired with the heavy chain is less than 10 and the average UMI count is less than or equal to 5, discard the corresponding light chain and its paired heavy chain types. Condition 2 - If the number of light chain types paired with the heavy chain is greater than 10 and the percentage is less than 50%, discard the corresponding light chain and its paired heavy chain types.
For contigs that contain only a light chain or a heavy chain in the barcode, contigs that meet either of the following conditions will be discarded: For BCR, if the contig's UMI is less than 3; for TCR, no such filtering will be applied.
If the total number of reads in the barcode containing a contig is less than or equal to 50, all contigs within that barcode will be discarded.
For contigs that appear in multiple barcodes and are present in over 75% of the barcodes, with each barcode containing either a light chain or a heavy chain only, contigs meeting both of the following conditions will be discarded: if their UMI is less than 5 or if their read count is less than half of the median read count across all barcode reads.
Filtering based on annotation results:
Based on the results from IgBLAST, only retain contigs that have a full sequence length greater than or equal to 300 bp and both 'complete_vdj' and 'productive' flags are set to TRUE.
Filtering for barcodes with excessive contigs:For each cell/barcode, considering the chain (e.g., TRA or TRB), a maximum of two sequences will be retained. If there are more than two, only the two contig sequences corresponding to the most frequently occurring CDR3 nucleotide sequences will be kept. In cases where each CDR3 nucleotide corresponds to multiple contig sequences, the contig sequence with the highest UMIs and read counts will be selected.
VDJ annotation
Annotation is performed using the IgBLAST software. For detailed information, please refer to https://ncbi.github.io/igblast/.
Reference sequences can be downloaded from the IMGT website (http://www.imgt.org), which includes five species: human, monkey, mouse, rat, and rabbit.
Cell identification
After sequence filtering through assembly, barcodes that contain one or more contigs are identified as cells.
Clonotype calculation
Clonotype calculation is performed using Change-O. For detailed information, please refer to https://changeo.readthedocs.io/en/stable/.