处理过程
step1: barcode/UMI提取
SeekSoulTools根据不同的Read1的结构设计和参数对barcode/UMI进行提取和处理,对Read1和Read2进行过滤,输出新的fastq文件。
结构设计和描述
barcode和UMI描述 以字母和数字描述Read1的基本结构,字母描述碱基含义,数字描述碱基长度。
B: barcode部分碱基
L: linker部分碱基
U: UMI部分碱基
X: 其他任意碱基,用于占位以下面两种Read1结构为例:
B8L8B8L10B8U8:
B17U12:

错位设计的Anchor定位 MM设计下,为了增加Linker部分在测序时碱基均衡性,加入了1-4 bp的移码碱基,即anchor,anchor决定了barcode的起始位置。

数据流程
确定anchor
对于Read1有错位设计的数据(MM试剂产出的数据),SeekSoulTools尝试在Read1序列前7个碱基中寻找anchor序列,以确定后续barcode等的起始。若没找到anchor序列,此条read以及对应的R2被认为是无效read。
Barcode和Linker矫正
在确定barcode的起始后,根据结构设计,取出相应序列。当取出的barcode序列在白名单中时,我们认为它是有效barcode,计入有效barcode的reads数量;当barcode不在白名单中时,我们认为它是无效barcode。
测序过程中,有一定几率发生测序错误。在提供有白名单的情况下,SeekSoulTools可以尝试barcode矫正。在启用矫正时,当无效barcode一个碱基错配(一个hamming distance)的序列存在于白名单中:
只有唯一一个序列存在于白名单中:我们将这个无效barcode矫正为白名单中barcode;
有多个序列存在于白名单中:我们将这个无效barcode改为read支持数量最多的序列;
Linker的处理与Barcode相同。
接头和PolyA序列剪切
在转录组产品中,Read2的末端有可能会出现polyA tail,建库时可能引入的接头序列。我们对这些污染序列进行切除,剪切完的read2长度需要大于设定的最小长度来保证有足够的信息,准确比对到基因组的位置。如果剪切完成后的read2长度小于最小长度,我们认为这条read为无效read。
经过上述处理后,数据组成如下图:
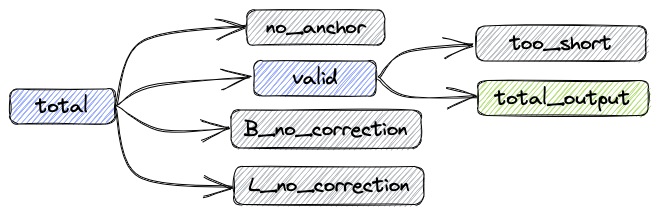
total: 总共的reads数目
valid: 不需要矫正和矫正成功的reads数目
B_corrected: 矫正成功的reads数目
B_no_correction: 错误Barcode的reads数目
L_no_correction: 错误Linker的reads数目
no_anchor:不包含anchor的reads数目
trimmed: 进行过剪切的reads数目
too_short: 进行过剪切后长度小于60bp的reads数目
指标之间的关系如下:
total = valid + no_anchor + B_no_correction + L_no_correction
输出fastq的reads数:total_output = valid - too_short
step2: 进行比对并找到比对基因
序列比对
使用STAR比对软件将处理后的R2比对到参考基因组上。
使用QualiMap软件和转录本注释文件GTF,统计reads比对外显子、内含子和基因间区等的比例。
使用featureCounts将比对上的read注释到基因上,可以选择不同的注释规则,如链方向性和定量的feature。当使用外显子定量时,当read超过50%碱基比对到外显子区域时,认为该read来源于此外显子以及外显子对应的基因;当使用转录本定量时,当read超过50%碱基比对到转录本区域时,认为该read来源于此转录本以及转录本对应的基因。
经过上述处理后,有如下数据指标:
Reads Mapped to Genome: 比对到参考基因组上的reads占所有reads的比例(包括只有一个比对位置和多个比对位置的reads)
Reads Mapped Confidently to Genome: 在参考基因组上只有一个比对位置的reads占所有reads的比例
Reads Mapped to Intergenic Regions:比对到基因间隔区reads占所有reads的比例
Reads Mapped to Intronic Regions:比对到内含子reads占所有reads的比例
Reads Mapped to Exonic Regions:比对到外显子reads占所有reads的比例
step3: 定量
UMI定量
SeekSoulTools以barcode为单位提取featureCounts输出的bam数据,统计注释到基因的UMI和UMI对应的reads数:
过滤掉对应UMI为单个重复碱基的reads, 例如UMI为TTTTTTTT
当read注释到多个基因,在有唯一外显子的注释时,为有效read,其他情形下都过滤掉
UMI矫正
测序过程中,UMI也有一定概几率出现测序错误。SeekSoulTools默认使用UMI-tools的adjacency方法对UMI进行矫正。
 Image source: https://umi-tools.readthedocs.io/en/latest/the_methods.html
Image source: https://umi-tools.readthedocs.io/en/latest/the_methods.html
细胞判定
在一个细胞群中,我们认为细胞和细胞的mRNA的含量不会相差太多。如果一个barcode对应的mRNA的含量很低,我们认为这个barcode的磁珠并没有捕获细胞,mRNA来源于背景。SeekSoulTools会以上面的规则,进行barcode是否为细胞的判定。有以下几个步骤:
对所有barcode按照对应的UMI数由高到低排序;
取预估细胞的1%位置的barcode的UMI数除以10为阈值;
barcode的UMI数大于阈值的判定为细胞;
barocde的UMI小于阈值,但大于300时,使用DropletUtils分析。DropletUtils方法先假设UMI数量低于100的barcode为empty droplet,然后根据每个droplet相同基因的UMI数总和为背景RNA表达谱中该基因UMI数目,进而得到基因UMI数目的期望值。再通过将每个barcode的UMI数进行统计学检验,显著差异的为细胞;
不符合上述条件的为背景。
经过上述处理后,有如下数据指标:
Estimated Number of Cells: 算法判定的细胞总数
Fraction Reads in Cells: 判定为细胞的reads占所有参与定量的reads的比例
Mean Reads per Cell: 细胞的平均reads数,总reads数/判定的细胞数
Median Genes per Cell: 判定为细胞的barcode中基因数的中位数
Median UMI Counts per Cell: 判定为细胞的barcode中UMI数的中位数
Total Genes Detected: 所有细胞检测到基因数量
Sequencing Saturation: 饱和度,1 - UMI总数/reads总数
step4: 后续分析
经过上述定量,得到表达矩阵后,我们可以进行下一步的分析。
Seurat分析流程
使用Seurat计算线粒体含量,细胞中UMI总数,细胞中基因总数。之后对矩阵进行归一化、寻找高变基因、降维聚类之后寻找差异基因。