处理过程
barcode/UMI提取
SeekSoulTools根据Read1的结构设计和参数对barcode/UMI进行提取和处理,对Read1和Read2进行过滤,输出新的fastq文件。
结构设计和描述
barcode和UMI描述 以字母和数字描述Read1的基本结构,字母描述碱基含义,数字描述碱基长度。
B: barcode部分碱基
U: UMI部分碱基
X: 其他任意碱基,用于占位
免疫分析产品的Read1结构为B17U12:

数据流程
Barcode识别和矫正
根据结构设计,确定barcode所在序列位置,取出相应序列。当取出的barcode序列在白名单中时,我们认为它是有效barcode,计入有效barcode的reads数量;当barcode不在白名单中时,我们认为它是无效barcode。
测序过程中,有一定几率发生测序错误。在提供有白名单的情况下,SeekSoulTools可以尝试barcode矫正。在启用矫正时,当无效barcode一个碱基错配(一个hamming distance)的序列存在于白名单中:
只有唯一一个序列存在于白名单中:我们将这个无效barcode矫正为白名单中barcode;
有多个序列存在于白名单中:我们将这个无效barcode改为read支持数量最多的序列;
接头和PolyA序列剪切
在建库时可能引入的接头或者TSO等序列,我们对这些污染序列进行切除,剪切完的read1和read2长度需要大于设定的最小长度来保证有足够的信息,准确比对到基因组的位置。如果剪切完成后的read1或read2长度小于最小长度,我们认为这条read为无效read。
经过上述处理后,数据指标包括:
total: 总共的reads数目
valid: 不需要矫正和矫正成功的reads数目
B_corrected: 矫正成功的reads数目
B_no_correction: 错误Barcode的reads数目
trimmed: 进行过剪切的reads数目
too_short: 进行过剪切后长度小于60bp的reads数目
输出fastq的reads数:total_output = valid - too_short
序列组装
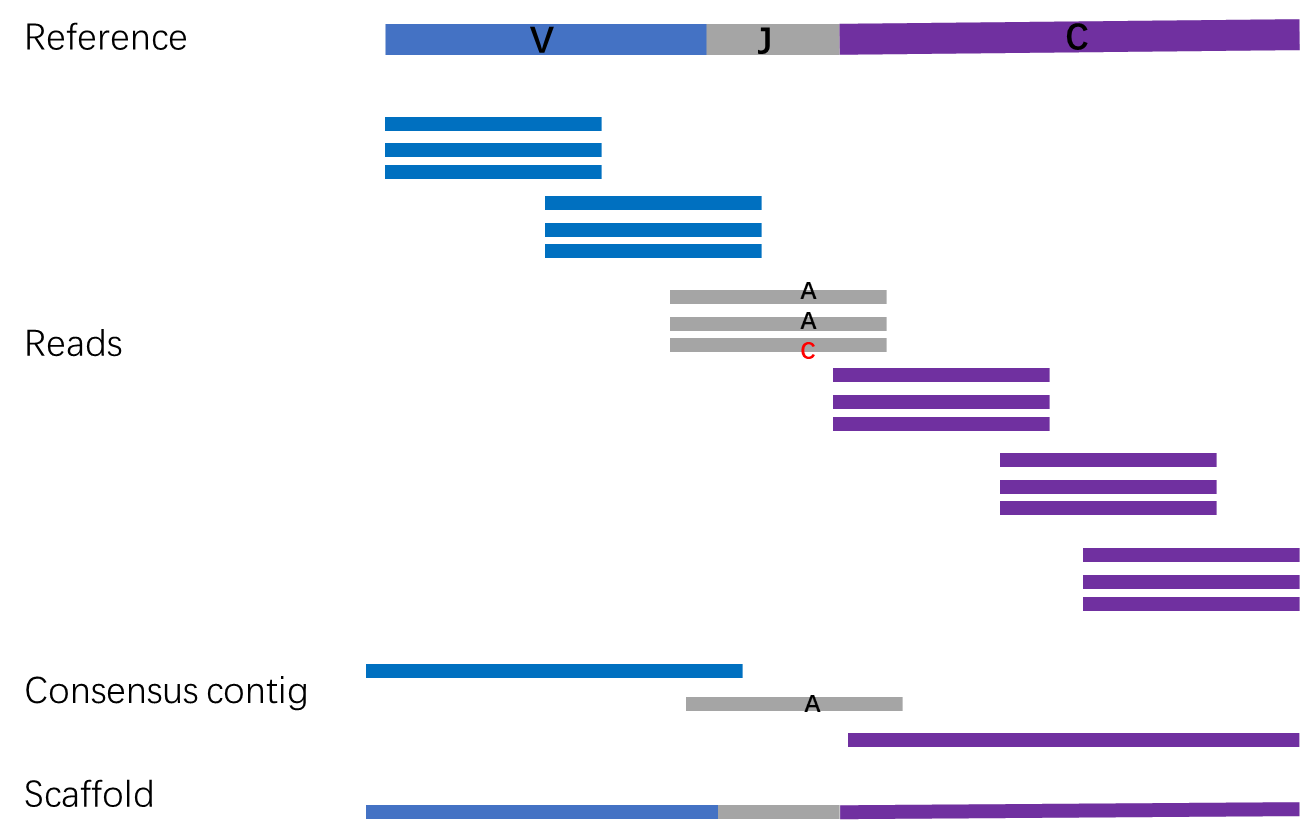
vdj序列组装指的是将测序reads装成为长的vdj序列,主要包含以下步骤(示意图如上):
将reads比对到参考序列,获得相对于参考序列的坐标。
首先,将read和VDJ的germline参考序列分别切成11bp的kmer,然后计算read和参考序列间共有的kmer数目,作为read与参考序列比对长度。接着,如果read和参考序列之间有22bp的碱基是完全匹配的,就认为该read可以比对到参考序列上。对于上述得到的可以比对到germline参考序列的一条read,我们根据比对长度,确认其参考序列是V-REGION,J-REGION或者C-REGION中的哪一种。一条read可以比对到V-REGION(J-REGION或者C-REGION)的多个基因。我们将同一barcode和同一umi的所有reads比对到最多的V-REGION(J-REGION或者C-REGION)的基因,作为最终的参考序列。最后,根据该参考序列,将read上的每个碱基,标记上相对于参考序列的坐标。
对具有相同参考序列坐标的reads上的碱基进行纠错。
分别计算对应于参考序列每个位置上的reads碱基的一致性。如果reads的覆盖深度大于或等于2,并且碱基的一致率大于或等于51%,则选取出现次数最多的碱基;反之则标记为N。
纠错后的reads,组装成长的consensus contig。
使用相对于V-REGION,J-REGION或者C-REGION的坐标,将reads之间进行连接,组装成长的consensus contig。 组装consensus contig的方法有两种(默认为使用第二种方式): 第一种,使用具有同一umi的reads进行组装,每个umi得到一个V-REGION,J-REGION或者C-REGION的contig; 第二种,将同一V-REGION(J-REGION或者C-REGION)基因的所有reads进行合并,分别得到一个V-REGION,J-REGION或者C-REGION的contig。
V-REGION,J-REGION和C-REGION的consensus contig之间的组装。
V-REGION,J-REGION和C-REGION的consensus contig之间通过overlap进行连接,形成完整的序列。将相互之间具有18 bp以上overlap的contig,进行连接。
组装序列过滤
根据组装指标进行过滤:
对于在barcode中同时存在轻链和重链的contig,则符合以下条件之一的contig会被丢掉:对于BCR,如果contig的umi小于3;对于TCR则不做该过滤。
理论上,在一个样本中,与同一种重链所匹配的轻链数量是有限的。而如果存在污染,则会出现一种重链对应很多种轻链的情况。对于umi小于或者等于10的contig,如果符合两个条件之一,会被过滤掉:条件1,在所有barcode中,与重链配对的轻链类型小于10种,并且平均umi数目小于或者等于5,则该轻链及其配对的重链类型。条件2,如果与重链配对的轻链大于10种,并且占比小于50%的轻链及其配对的重链类型。
对于在barcode中仅存在轻链或者重链的contig,则符合以下条件之一的contig会被丢掉:对于BCR,如果contig的umi小于3;对于TCR则不做该过滤。
contig所在的barcode的总reads数目小于或者等于50,则该barcode中的所有contig均丢掉。
对于在多个barcode中出现,并且在超过75%的barcode中都只包含轻链或者重链的,同时umi小于5或者reads小于所有barcodereads数中值1/2的contig。
根据注释结果进行过滤:
根据igblast的结果,仅保留序列全长大于或等于300 bp,并且complete_vdj和productive均为TRUE的contig。
对一个barcode有过多的contig的进行过滤: 对于每个细胞/barcode中chain(例如TRA或者TRB)来讲,最多保留两个序列。如果超出两个,则仅保留出现次数最多的两个CDR3核酸序列对应的组装序列。如果每个CDR3核酸对应多个组装序列,则选取umis和reads数目最多的组装序列。
VDJ注释
注释使用igblast软件,详细请参考https://ncbi.github.io/igblast/。
参考序列下载子IMGT网站(http://www.imgt.org),包含人,猴,小鼠,大鼠和兔等五个物种。
细胞鉴定
经组装序列过滤后,包含一个及以上contig的barcode,判定为细胞。
clonotype计算
clonotype计算使用changeo,详细请参考https://changeo.readthedocs.io/en/stable/。